Синдром марфана
Содержание:
- Осложнения и риски синдрома Марфана
- Диагностика
- 2.Наследственные заболевания соединительной ткани
- Патогенез (что происходит?) во время Поражений легких при синдроме марфана:
- Marfan syndrome causes
- Прогноз и профилактика
- Патологическая анатомия
- Диагностика
- Проявления заболевания
- Синдром Марфана
- Мутации в гене » FBN1 »
- Гиперсексуальность
- Симптомы синдрома Марфана
- Marfan syndrome complications
- Симптоматика
- Развитие сердечно-сосудистой системы плода [ править ]
- Близорукость
Осложнения и риски синдрома Марфана
Синдром Марфана приводит к …
По мере того как органы будут затрагиваться в ходе развития синдрома Марфана, могут появиться различные осложнения, в частности:
- Аневризма и рассечение аорты, как восходящей, так и брюшной. Диссекция аорты часто отмечается во время беременности. В этом состоянии сердце матери подвергается чрезмерному напряжению, что может оказаться фатальным для целостности аорты.
- Нарушения митрального и/или аортального клапана. В результате того, что сердце „переутомляется“, что неизбежно ведет к сердечной недостаточности.
- Отслоение сетчатки.
- Вывих хрусталика и его дислокация.
- Возникновение ранней катаракты и глаукомы.
- Деформация скелета (грудина, позвоночник, ноги).
Диагностика
УЗИ человека с синдромом Марфана, показывающее расширенный корень аорты
Диагностические критерии MFS были согласованы на международном уровне в 1996 году. Однако синдром Марфана часто трудно диагностировать у детей, поскольку они обычно не проявляют симптомов до достижения полового созревания. Диагноз основывается на семейном анамнезе и комбинации основных и второстепенных показателей расстройства, редко встречающихся в общей популяции, которые встречаются у одного человека, например: четыре скелетных признака с одним или несколькими признаками в другой системе организма, такой как глазная и сердечно-сосудистые у одного человека. Следующие состояния могут быть результатом MFS, но могут также возникать у людей без каких-либо известных основных заболеваний.
- Аневризма или расширение аорты
- Арахнодактилия
- ГЭРБ
- Двустворчатый аортальный клапан
- Кисты
- Кистозный медиальный некроз
- Дегенеративная болезнь диска
- Искривленная перегородка
- Дуральная эктазия
- Ранняя катаракта
- Ранняя глаукома
- Ранний остеоартроз
- Эктопия лентис
- Эмфизема
- Ирис колобома
- Выше среднего роста
- Учащенное сердцебиение
- Грыжи
- Небо с высокой аркой
- Гипермобильность суставов
- Кифоз (сгорбившись)
- Негерметичный сердечный клапан
- Неправильный прикус
- Микрогнатия (малая нижняя челюсть)
- Пролапс митрального клапана
- Близорукость (близорукость)
- Обструктивная болезнь легких
- Остеопения (низкая плотность костной ткани)
- Pectus carinatum или экскаватум
- Pes planus ( плоскостопие )
- Пневмоторакс (коллапс легкого)
- Отслойка сетчатки
- Сколиоз
- Апноэ во сне
- Растяжки не от беременности или ожирения
- Зубы скучены
- «Узкое, худое лицо»
- Дисфункция височно-нижнечелюстного сустава (ВНЧС)
Пересмотренная гентская нозология
Знак большого пальца; верхний : нормальный, нижний : синдром Марфана
В 2010 году нозология Гента была пересмотрена, и новые диагностические критерии заменили предыдущее соглашение, заключенное в 1996 году. Семь новых критериев могут привести к постановке диагноза:
При отсутствии семейного анамнеза MFS:
- Z-показатель корня аорты ≥ 2 И эктопия lentis
- Z-показатель корня аорты ≥ 2 И мутация FBN1
- Z-оценка корня аорты ≥ 2 И системная оценка *> 7 баллов
- Ectopia lentis И мутация FBN1 с известной патологией аорты
При наличии семейного анамнеза MFS (как определено выше):
- Эктопия лентис
- Системный балл * ≥ 7
- Z-показатель корня аорты ≥ 2
- Очки за системный балл:
- Знак запястья И большого пальца = 3 (знак запястья ИЛИ большого пальца = 1)
- Деформация Pectus carinatum = 2 (асимметрия грудной клетки или грудной клетки = 1)
- Деформация заднего отдела стопы = 2 (плоская стопа = 1)
- Эктазия твердой мозговой оболочки = 2
- Протрузия вертлужной впадины = 2
- пневмоторакс = 2
- Уменьшение соотношения верхнего и нижнего сегментов И увеличение руки / роста И отсутствие тяжелого сколиоза = 1
- Сколиоз или грудопоясничный кифоз = 1
- Уменьшенное разгибание локтей = 1
- Черты лица (3/5) = 1 ( долихоцефалия , энофтальм , опускание глазных щелей , гипоплазия скуловой кости , ретрогнатия )
- Бороздки кожи ( растяжки ) = 1
- Близорукость > 3 диоптрии = 1
- Пролапс митрального клапана = 1
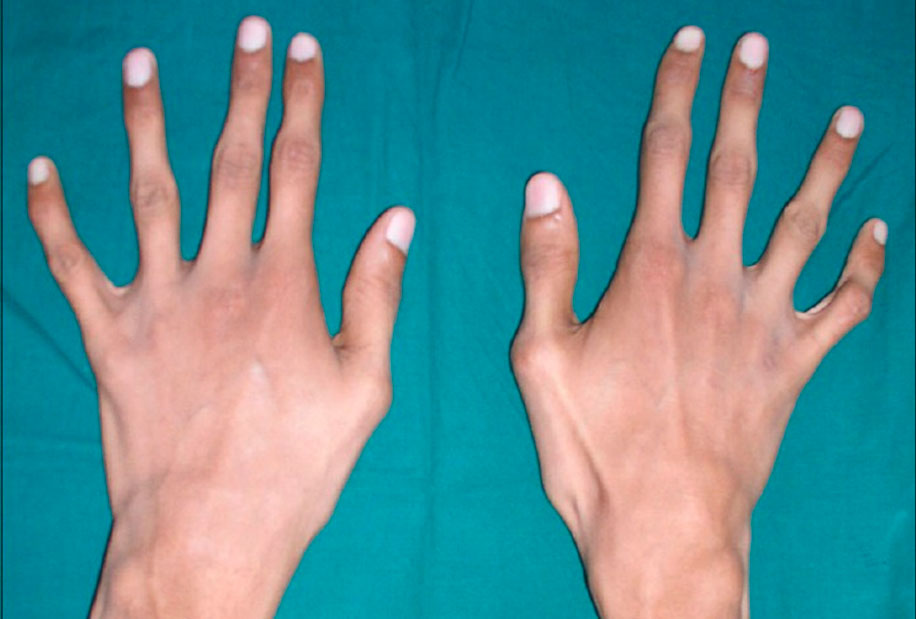
Знак большого пальца (знак Стейнберга) вызывается просьбой человека согнуть большой палец как можно дальше, а затем сомкнуть пальцы над ним. Положительный признак большого пальца — это когда вся дистальная фаланга видна за локтевым краем кисти, что вызвано сочетанием гипермобильности большого пальца, а также большого пальца, который длиннее обычного.
Знак запястья (знак Уокера-Мердока) вызывается, когда человека просят обхватить большим пальцем и пальцами одной руки другое запястье. Положительным признаком запястья является перекрытие мизинца и большого пальца из-за сочетания тонких запястий и длинных пальцев.
Дифференциальная диагностика
Многие другие расстройства могут вызывать те же характеристики тела, что и синдром Марфана. Генетическое тестирование и оценка других признаков и симптомов могут помочь их дифференцировать. Ниже приведены некоторые расстройства, которые могут проявляться как «марфаноид»:
- Врожденная контрактурная арахнодактилия , также известная как синдром Билса-Хехта
- Синдром Элерса-Данлоса
- Гомоцистинурия
- Синдром Лойса-Дитца
- МАСС-фенотип
- Множественная эндокринная неоплазия 2В типа
- Синдром Шпринцена – Гольдберга
- Синдром Стиклера
2.Наследственные заболевания соединительной ткани
Некоторые болезни соединительной ткани являются результатом изменений в определенных генах. Вот наиболее распространенные из них:
Синдром Элерса-Данло
Данный синдром представляет собой группу заболеваний наследственного характера, отличительной особенностью которых являются гипермягкие суставы, повышенная эластичность кожи, подверженные повреждениям кровеносные сосуды, аномальный рост рубцовой ткани и синяки. Симптоматика данного синдрома может варьироваться от легкой степени до высокой. В зависимости от конкретного вида Элерса-Данло признаки этого заболевания могут включать следующее:
- кровоточивость десен,
- слабые сосуды,
- медленное заживление ран,
- изогнутый позвоночник,
- плоскостопие,
- проблемы с легкими, сердечными клапанами и органами пищеварения.
Врожденный буллезный эпидермолиз
Люди с данным заболеванием имеют настолько хрупкую кожу, что практически любое соприкосновение с ней сопровождается возникновением тонкостенных волдырей с серозным содержимым. Подобные пузыри могут возникнуть при ударе, падении человека на землю и даже от трения одежды на определенных участках кожи.
В зависимости от какого-либо конкретного вида врожденного буллезного эпидермолиза подвергаться неблагоприятному воздействию могут дыхательные пути, пищеварительный тракт, мочевой пузырь, мышцы или другие органы. Как правило, буллезный эпидермолиз проявляется сразу при рождении ребенка, так как сам акт родов в некоторой степени является первой механической травмой.
Синдром Марфана
Клиническая картина данного заболевания характеризуется поражением большинства жизненно важных органов и систем: сердечно-сосудистой и центральной нервной системы, опорно-двигательного аппарата, органов зрения и дыхания. Люди с синдромом Марфана имеет высокий рост, а также чрезмерно длинные кости и тонкие «паукообразные» пальцы ног, рук.
Среди других проблем, сопровождающих синдром Марфана, можно выделить проблемы со зрением по причине аномального размещения хрусталика глаза и расширения аорты. Синдром Марфана провоцируется мутациями в гене, который регулирует структуру белка фибриллина.
Несовершенный остеогенез
Данная патология является врожденным расстройством, характеризующимся хрупкостью костей, низкой мышечной массой. Существует несколько типов этого заболевания. Специфические симптомы несовершенного остеогенеза зависят от конкретного вида заболевания и могут включать в себя следующие признаки:
- серый или синий оттенок склеры – белка глаз;
- тонкая кожа;
- снижение слуха;
- незначительное искривление позвоночника;
- проблемы с дыханием.
Причина возникновения данной патологии заключается в мутации генов COL1A1 и COL1A2, ответственных за коллаген типа 1, а также изменение качества протеина.
Патогенез (что происходит?) во время Поражений легких при синдроме марфана:
Патогенез заболевания неясен. По-видимому, мутация, определяющая это заболевание, касается гена, кодирующего структурный белок, входящий в состав коллагена. В процессе роста значительное количество коллагеновых волокон развивается не полностью или дегенерирует. Вследствие этого возникают поражения опорно-двигательного аппарата, глаз и внутренних органов, в том числе легких.
Гистологически отмечаются фрагментация, расщепление, истончение, местами полное исчезновение эластических волокон соединительной ткани, наличие замещающей их фиброзной ткани, богатой тучными клетками с явлениями мукоидизации и ме-тахромазии. Характерно накопление в организме свободных или слабосвязанных с белком кислых мукополисахаридов (гликоза-миногликанов). Отмечается дисфункция коркового вещества надпочечников, проявляющаяся в нарушении суточного ритма экскреции глюкокортикоидов, соотношения между кортикостероном и гидрокортизоном в крови.
Marfan syndrome causes
Marfan syndrome is caused by a gene abnormality, specifically a change (mutation) in the FBN1 gene that affects the elasticity of connective tissue in muscles and joints. The FBN1 gene provides instructions for making a protein called fibrillin-1. Fibrillin-1 attaches (binds) to other fibrillin-1 proteins and other molecules to form threadlike filaments called microfibrils. Microfibrils become part of the fibers that provide strength and flexibility to connective tissue. Additionally, microfibrils store molecules called growth factors and release them at various times to control the growth and repair of tissues and organs throughout the body. A mutation in the FBN1 gene can reduce the amount of functional fibrillin-1 that is available to form microfibrils, which leads to decreased microfibril formation. As a result, excess growth factors are released and elasticity in many tissues is decreased, leading to overgrowth and instability of tissues.
- Marfan syndrome is most often inherited from a parent in an autosomal dominant pattern, who will have a 50% chance of passing the condition on to their children.
- However, in about one quarter of people diagnosed with Marfan syndrome, nobody else in the family is affected – the disease is due to a new mutation in the FBN1 gene. These cases occur in people with no history of the disorder in their family.
The FBN1 gene is in the long arm of chromosome 15 at position 21.1 (15q21.1).
Прогноз и профилактика
Больным с синдромом Марфана необходимо комплексное лечение всех патологий. Если такое лечение не проводится, то продолжительность жизни заболевших редко превышает 40 лет. Чаще всего к этому возрасту нарастает тяжелая сердечная недостаточность, могут возникнуть патологии сосудов, обширные кровоизлияния и смерть. Однако если проводить регулярное обследование и лечение сопутствующих патологий, люди с синдромом могут прожить долгую жизнь.
Особого внимания требует к себе женщина с синдромом Марфана во время беременности. Если проводить регулярное и комплексное обследование и принимать препараты, препятствующие ухудшению состояния сердечнососудистой системы, то беременность протекает успешно и заканчивается рождением ребенка.
Нет способов предотвратить развитие патологии, поскольку болезнь носит генетическую природу. Если синдром Марфана есть у одного из родителей, высока вероятность того, что заболевание разовьется и у их ребенка, поскольку для него характерный аутосомно-доминантный тип наследования.
Люди с синдромом Марфана должны проводить регулярную профилактику заболеваний сердечнососудистой и опорно-двигательной системы. Для этих целей рекомендуется вести здоровый образ жизни и регулярно проводит диагностические обследования. Для детей не рекомендована усиленная физическая нагрузка, однако им следует регулярно заниматься лечебной физкультурой.
Патологическая анатомия
При морфологическом исследовании — эластические волокна истончены, расположены неравномерно, местами хаотично; наблюдается расслоение средней оболочки крупных сосудов, разрыхление эндотелиального слоя, образование в эндотелиальном и субэндотелиальном слоях подушкообразных выступов в просвет сосуда (см. Аневризма расслаивающая). Эластический каркас аорты и легочного ствола развит слабо. Миокард с дистрофическими изменениями, вакуолизацией, местами с резким набуханием волокон. В костной ткани наблюдается разреженность костных балок и неравномерное отложение извести; нарушена структура хрящевой ткани за счет образования коллагеновых пучков, расслаивающих межуточное вещество.
Диагностика
Диагноз синдром Марфана ставится на основании имеющейся клинической картины, семейного анамнеза и генетических исследований.
Процессу диагностики помогает ряд визуальных исследований. Рентгенография дает информацию о состоянии грудной клетки, изменении длины и ширины костей ладоней и пальцев. Рентгенограммы таза отражают состояние тазобедренных суставов. КТ и МРТ имеют преимущества в оценке состояния мягких тканей и детализации дефектов.
Сердечно-сосудистая система оценивается на основании физического осмотра, касающегося дефекта клапана, признаков сердечной недостаточности, ЭКГ и обычной, а в некоторых случаях чреспищеводной эхокардиографии. Состояние аорты и крупных сосудов оценивают контрастным исследованием.
Исследование зрительной системы начинается с определения остроты зрения, офтальмоскопии, осмотра глазного дна. Широко распространенным исследованием в последнее время является УЗИ глазного яблока.
Проявления заболевания
Данной патологии сопутствует множество нарушений в организме. Так называемая «триада Марфана» — это повреждение скелетных костей, болезни сердечно-сосудистой системы и различные нарушения зрения. Однако надо отметить, что на умственные способности этот синдром никак не влияет.
Заболевшие отличаются, как правило, непропорциональным сложением: при довольно высоком росте у них укороченное туловище, длинные руки и пальцы, напоминающие паучьи (арахнодактилия), астеническое телосложение с мышечной гипотонией, деформация грудной клетки, сильное искривление позвоночника (кифоз, сколиоз, подвывихи и вывихи шейного отдела), плоскостопие.
Синдром Марфана, сопровождающийся аномалией сердечно-сосудистой системы, выражается в дефектах стенок крупных сосудов, особенно аорты и больших ветвей легочных сосудов, пороками развития сердца. Эти изменения часто формируются уже у развивающегося плода. Самая опасная форма синдрома Марфана, симптомы которой проявляются уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни младенца.
Синдром Марфана
Синдром Марфана (MFS) представляет собой аутосомно-доминантное заболевание, которое поражает соединительные ткани систем организма, таких как глаза, сердечно-сосудистая система, скелетная система, кожа, легочная система и твердая мозговая оболочка. MFS поражает примерно 1 из 5000 человек. MFS не является легко диагностируемой патологией с использованием балльной системы, называемой нозологической таблицей Гента, а не тестом на одну молекулу. Чтобы диагностировать пациентов с MFS, у которых не было семейной истории, необходимо соответствие двум критериям. Во-первых, должны быть затронуты две разные основные системы органов, а во-вторых, должна быть задействована третья система органов.
MFS обычно возникает из-за мутаций De Novo и приводит к индивидуальному фенотипическому проявлению длинных и тонких конечностей и конечностей, изогнутых позвоночников, обычно приводящих к сколиозу грудного отдела, гипергибким суставам, экскаватору грудной клетки, отслоению сетчатки и впалой груди. Мутации «De Novo», приводящие к тяжелому MFS, имеют высокий ожидаемый уровень смертности новорожденных. Классические симптомы MFS обычно становятся заметными в период полового созревания или в более позднем возрасте; редко развивается на ранних этапах жизни. Наиболее частым кожным проявлением MFS являются растяжки, когда полосы кожи окрашиваются в красный, фиолетовый и затем белый цвета. Эпидермис кожи тонкий и уплощенный, а толщина верхнего защитного слоя кожи уменьшена. Это проявление гистологически характеризуется прямыми тонкими пучками коллагена, расположенными параллельно коже и эластическим волокнам. Эластичные волокна более плотные в верхней части дермы, а под этой зоной имеется локальное отсутствие эластичных волокон. Между границами стрий и кожей иногда присутствуют загнутые, оборванные ретикулярные эластические волокна. Эти симптомы являются причиной появления паутины на коже у пациентов с MFS.
Лечение MFS заключается в оперировании человека посредством операции на открытом сердце. Ведение MFS включает стандартные последствия, такие как консультирование по образу жизни для снижения и ограничения физической активности, эндопрофилактика, серийная визуализация аорты, прием ß-блокаторов для защиты аорты и профилактическое замещение корня аорты. Взрослым, страдающим MFS, рекомендуется снизить эмоциональное и физическое напряжение и переключиться с высокоэффективных видов спорта, таких как боевые искусства, футбол, баскетбол и т. Д., На изотонические упражнения с малой ударной нагрузкой, такие как плавание, езда на велосипеде или бег трусцой, при которых частота пульса составляет примерно Дети также должны следовать аналогичным рекомендациям, чтобы обеспечить правильное ведение MFS.
MFS вызывается мутацией в гене «’ FBN1 ’’, расположенным на хромосоме 15q21.1, в результате чего образуется деконструированная форма фибриллина-1. Фибриллин-1 представляет собой богатый цистином гликопротеин массой 350 кДа и 2871 аминокислот, который отвечает за слияние эластина с эластичными волокнами соединительной ткани во внеклеточном матриксе (ЕСМ).Хрупкость соединительной ткани обычно приводит к аневризмам аорты из-за того, что стенка неспособна выдерживать внутрипросветное давление. Дефекты фибриллина-1 приводят к повышенным уровням TGF-β, которые напрямую коррелируют с MFS.
Мутации в гене » FBN1 »
«’ FBN-1 ’’ представляет собой ген размером примерно 200 т.п.н. и состоит из большой кодирующей последовательности, разделенной на 65 экзонов, расположенных на хромосоме 15. Этот ген кодирует белок фибриллин-1. Фибриллин-1 представляет собой большой гликопротеин, богатый цистеином, приблизительно 350 кДа, в основном состоящий из тандемно повторяющихся доменов модулей, подобных эпидермальному фактору роста (ECF). Эти домены гомологичны кальций-связывающему модулю эпидермального фактора роста (cbEGF-подобные мотивы) и отдельным 8-цистеиновым модулям, составляющим эластичную и неэластичную ткань. Эти эластичные и неэластичные ткани представляют собой микрофибриллярные пучки, гетерополимеры как фибриллина-1, так и фибриллина-2. Эластогенез — это биологический процесс, при котором микрофибриллы и эластичные волокна самоорганизуются посредством организованного осаждения несколькими макромолекулами. Полимеризованные фибриллины могут быть охарактеризованы их структурой микрофибрилл «шарики на нитке»; образуя решетку микрофибрилл через боковую связь отдельных полимеров и структурных компонентов.
Мутации фибриллина-1 являются основным мутировавшим белком, вызывающим MFS. Эта мутация обычно мешает сборке микрофибрилл, что приводит к доминантно-отрицательному механизму.
Мутации могут включать:
- Миссенс-мутации, вызванные заменами одного основания, такого как цистеин, или тех, которые связаны со связыванием кальция в фибриллине-1.
- Преждевременное прерывание из-за бессмысленных мутаций или сдвига рамки считывания.
- Мутации в экзонном сайте сплайсинга, допускающие вставки или удаления из-за создания загадочных сайтов сплайсинга.
- Изменения в основании сайта интронного сплайсинга, приводящие к альтернативному сплайсингу и пропуску или удалению экзона в рамке.
Комбинация четырех типов мутаций приводит к неправильной экспрессии фибриллина-1. Нет корреляции между фенотипом и генотипом на молекулярном уровне.
Известны мутации гена FBN-1 в шести хромосомных локусах, TAAD1 в 5q13-14, FAA1 в 11q23-24, TAAD2 в 3p24-25, TAAD3 в 15q24-26, TAAD4 в 10q23-24 и MYH11 в 16p12-13. быть триггерами MFS. Эти локусы, как правило, содержат гены, участвующие в сосудистой функции. Ген MYH11 отвечает за тяжелую цепь миозина гладких мышц, а ACTA2 в локусах TAAD4 кодирует альфа-актин гладких мышц.
Несинонимичная замена аминокислоты, затрагивающая консервативный цистеин в CaB-EGF-подобном домене, кодируемом экзоном 13 гена FBN1, может вызвать развитие MFS. Более высокая частота и серьезность MFS возникает, когда есть неправильные замены в дисульфидных связях C1-C2 или C3-C4, поэтому правильная локализация цистеина и дисульфидные связи в этих локусах имеют решающее значение для структурной целостности. Мутации в гене FBN1, приводящие к неправильному связыванию дисульфидной связи C5-C6, обычно приводят к MFS меньшей степени тяжести. Концентрированные мутации домена CaB-EGF вдоль полипептида FBN1 влияют на фенотип тяжести MFS. Мутации с локализованной заменой цистеина в C538P в экзоне 13, C570R в экзоне 14 или C587Y в экзоне 15 приводят к симптомам MFS, связанным с глазами, в частности эктопией lentis. Сами микрофибриллы могут поддерживать гемодинамическую нагрузку в кровеносных системах беспозвоночных и мелких позвоночных. Эластин и развитие системы ECM, интегрированной с окружающим VSMC, необходимы для правильного функционирования высших позвоночных. Фибриллин-1 важен не для стабилизации эластичного элемента, а для сборки микрофибриллы. Повышающая регуляция активина А работает вместе с сигнальными молекулами фибриллина-1 и TGF-ß, вызывая фибропролиферативный ответ. Индукция CYR61 также поддерживает клеточную адгезию и регулирует ремоделирование матрикса и играет основную роль в формировании крупных сосудов и их целостности.
Гиперсексуальность
Синдромы Марфана и Морриса исключительно редки. Но для раскрытия механизмов гениальности оба синдрома необычайно важны. Синдром Марфана раскрывает значение адреналинового стимула, который имеется, пусть не в столь выраженном виде, у множества людей, этим синдромом не страдающих. Редчайший синдром Морриса показывает действие избыточных свободных андрогенов, также присутствующих у многих лиц, свободных от этого синдрома.
Синдромы Марфана и Морриса «прокладывают путь» к прочим, гораздо более частым, гормональным механизмам гениальности. Например, особое место занимает необычайная энергия и сила воли, порожденная сексуальным воздержанием (одним из примеров является монах Гильдебранд — папа Григорий VII) или, может быть, высокой сексуальностью.
Безудержной сексуальностью отличались Генрих II Плантагенет, Ярослав Мудрый, Атилла, Чингиз-хан, Гаральд Гарфагар, Иван Грозный, Август II Сильный, Генрих VIII Тюдор, Генрих IV Бурбон, Петр I, Екатерина II, Жорж Санд, Д.Г. Байрон, А.С. Пушкин, М.Ю. Лермонтов, Ф.И. Тютчев, А. Мюссе, О. Бальзак, Г. Гейне, Г. Мопассан, А.А. Блок, С.А. Есенин, В.В. Маяковский, Л.Н. Толстой и многие другие. У некоторых повышенная сексуальность сохранялась до глубокой старости (И.В. Гете), и если биографии многих замечательных деятелей свидетельствуют о полной или почти полной сублимации секса творчеством (Кант, Людвиг ван Бетховен), то когда-нибудь появится возможность рассмотреть и этот возможный источник творческой потенции, столь ярко проявившийся у аскетов-подвижников, священников, монахов.
Вопрос о том, играет ли повышенный уровень мужских гормонов у мужчин роль такого же мощного внутреннего допинга, как при синдроме Морриса у псевдогермафродитов, необычайно сложен и, конечно, решается неоднозначно. «Половые железы… интенсифицируют все физиологические, умственные и духовные виды активности. Ни один евнух не стал великим философом, великим ученым или великим преступником. Семенники повышают смелость, агрессивность и жестокость, качества, отличающие быка-драчуна от вола, тащащего плуг по полю» (А. Carrel).
Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов; — аномалии строения тазобедренного сустава; — кифоз, сколиоз; — вывихи шейного сегмента позвоночника; — деформация грудной клетки; — плоскостопие; — глубокая посадка глаз; — уменьшенная нижняя челюсть, нарушение роста зубов; — высокое нёбо; — атрофические «растяжки» на коже; — паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения); — пороки сердца (чаще — поражения клапанов); — стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Marfan syndrome complications
Because Marfan syndrome can affect almost any part of your body, it may cause a wide variety of complications.
Cardiovascular complications
The most dangerous complications of Marfan syndrome involve the heart and blood vessels. Faulty connective tissue can weaken the aorta — the large artery that arises from the heart and supplies blood to the body.
- Aortic aneurysm. The pressure of blood leaving your heart can cause the wall of your aorta to bulge out, like a weak spot in a tire. In people who have Marfan syndrome, this is most likely to happen at the aortic root — where the artery leaves your heart.
- Aortic dissection. The wall of the aorta is made up of layers. Dissection occurs when a small tear in the innermost layer of the aorta’s wall allows blood to squeeze in between the inner and outer layers of the wall. This can cause severe pain in the chest or back. An aortic dissection weakens the vessel’s structure and can result in a rupture, which may be fatal.
- Valve malformations. People who have Marfan syndrome also are more likely to have problems with their heart valves, which may be malformed or overly elastic. When heart valves don’t work properly, your heart often has to work harder to compensate. This can eventually lead to heart failure.
Eye complications
Eye complications may include:
- Lens dislocation. The focusing lens within your eye can move out of place if its supporting structures weaken. The medical term for this problem is ectopia lentis, and it occurs in more than half the people who have Marfan syndrome.
- Retinal problems. Marfan syndrome also increases the risk of a detachment or tear in the retina, the light-sensitive tissue that lines the back wall of your eye.
- Early-onset glaucoma or cataracts. People who have Marfan syndrome tend to develop these eye problems at a younger age. Glaucoma causes the pressure within the eye to increase, which can damage the optic nerve. Cataracts are cloudy areas in the eye’s normally clear lens.
Skeletal complications
Marfan syndrome increases the risk of abnormal curves in the spine, such as scoliosis. It can also interfere with the normal development of the ribs, which can cause the breastbone to either protrude or appear sunken into the chest. Foot pain and low back pain are common with Marfan syndrome.
Complications of pregnancy
Marfan syndrome can weaken the walls of the aorta, the main artery that leaves the heart. During pregnancy, a woman’s heart is pumping more blood than usual, and this can put extra stress on a woman’s aorta — which increases the risk of a deadly dissection or rupture.
Симптоматика
Синдром Марфана проявляется разнообразными клиническими признаками, что связано с присутствием соединительнотканных волокон в различных структурах организма. У больных появляются признаки поражения костей и суставов, зрительного анализатора, сердца, сосудов, нервов, легких, кожного покрова.
Симптомы дисфункциональных расстройств внутренних органов возникают на фоне сильной усталости и быстрой утомляемости после незначительных физических нагрузок, миалгии, вялости, мышечной гипотонии, приступообразной цефалгии. По мере роста и развития организма человека черты заболевания становятся более выраженными.
Скелет
Клинические признаки синдрома Марфана, обусловленные поражением костей, мышц и связок:
- короткое туловище и длинные ноги,
- молоткообразные пальцы стопы,
- паучьи пальцы кисти,
- худощавое телосложение,
- гипотонус мышц,
- удлиненное и узковатое лицо,
- глубокая посадка глаз,
- отсутствие зазоров между зубами,
- большой нос,
- микрогнатия,
- недоразвитые скулы,
- большие и низко расположенные уши,
- «готическое» небо,
- прогения,
- гиперподвижность суставов,
- деформированная грудная клетка в виде воронки или киля птицы,
- искривление позвоночника,
- смещение вышележащего позвонка относительно нижележащего,
- уплощение сводов стопы,
- продавливание вертлужной впадины.
Сердце и сосуды
Поражение сердечной мышцы при синдроме Марфана определяет его исход. Обычно возникают дефекты в структуре аорты, легочного ствола, развиваются пороки развития клапанов сердца:
- дилатация предсердий и желудочков,
- мешкообразное расширение ограниченного участка аортальной стенки,
- пролабирование двустворчатого клапана,
- миксоматоз сердца,
- кардиомиопатия,
- разрыв хорд митрального клапана,
- кальциноз аортального клапана,
- различные виды аритмии — мерцательная, экстрасистолия,
- воспаление внутренней оболочки сердца — эндокарда,
- кардиалгия с иррадиацией в спину, ключицу, руку, плечо,
- похолодание конечностей,
- затрудненное дыхание,
- сердечные шумы,
- на ЭКГ — признаки ИБС.
Глаза
У лиц с синдромом Марфана развивается патология зрения:
- миопия,
- смещение хрусталика в сторону,
- уплощение роговой оболочки глаза и ее утолщение,
- недоразвитие радужки и ресничной мышцы,
-
страбизм,
- катаракта,
- асимметричность зрачков,
- глаукома,
- астигматизм,
- дальнозоркость,
- колобома глаза,
- спазмирование сосудов сетчатки и высокий риск ее отслойки.
Прочие органы
- ишемические и геморрагические инсульты,
- субарахноидальное кровотечение,
- выпячивание оболочек спинного мозга.
Ослабление и растяжение дурального мешка проявляется болью в пояснице, ногах, тазу и другими неврологическими признаками, цефалгией.
- скопление воздуха между висцеральным и париетальным листками плевры,
- эмфизема легких,
- удлинение и перерастяжение альвеол,
- ночное апноэ,
- рак легких.
Поражение кожи, мягких тканей, внутренних органов:
- смещение почки в каудальном направлении,
- цистоцеле,
- пролапс матки,
- кисты и новообразования в печени и почках,
- варикоз,
- стрии на коже,
- грыжи.
Больные с синдромом Марфана легко возбудимы, гиперактивны, чрезмерно эмоциональны, часто плаксивы и разражительны. Они имеют неординарные способности и высокий уровень интеллекта.
«Готическое» небо и микрогнатия приводят к возникновению речевых нарушений. При поражении скелета и суставов появляются артралгии и миалгии, развивается ранний остеоартрит.
Синдром Марфана проявляется по-разному у лиц, обращающихся за медицинской помощью. У одних выявляются ярко выраженные признаки патологии, у других симптоматика бывает стертой. Прогрессирование синдрома происходит по мере взросления человека. Клинические проявления определяются локализацией и интенсивностью поражения органов и систем.
Развитие сердечно-сосудистой системы плода [ править ]
Ген FBN-1 участвует во множестве программ эмбрионального развития. Микрофибриллы, состоящие из фибриллина-1, способствуют формированию как эластичных, так и неэластичных структур. Формирование эластических волокон в сердечных клапанах и аорте требует участия как FBN-1, так и FBN-2. Было показано, что как FBN-1, так и FBN-2, наряду с другими компонентами эластических волокон, экспрессируются в эмбриональных полулунных клапанах уже на 4 неделе беременности. Эти молекулы взаимодействуют с образованием эластичных волокон в желудочковом слое полулунных клапанов. Фибриллин-1 и фибриллин-2 также имеют решающее значение для развития эластических волокон в аорте. Хотя экспрессия фибриллина-2 значительно снижается после внутриутробного развития плода, экспрессия фибриллина-1 продолжается и во взрослой жизни. Это подтверждает идею о том, что фибрилин-2 диктует развитие ранних эластических волокон, тогда как фибриллин-1 обеспечивает структурную поддержку зрелых эластических волокон.
Когда происходят мутации в генах FBN-1 или FBN-2, значительные деформации могут возникнуть в результате повреждения внеклеточного матрикса. Синдром Марфана — врожденное заболевание, возникающее в результате мутации гена FBN-1. Это приводит к уродству и последующему ослаблению микрофибрилл в организме пациента, в том числе в структурах сердечно-сосудистой системы. Ослабленные эластичные волокна приведут к нарушению прочности и растяжимости сердечных клапанов и аорты. Это объясняет аневризмы аорты и выпадение клапанов, которые обычно связаны с синдромом Марфана.
Близорукость
Данное нарушение зрения проявляется более чем у половины больных. Причинами ее могут быть: шаровидная форма хрусталика, изменение преломляющей силы роговицы глаза вследствие ее уплощения и увеличения, а также быстрый рост по переднезадней оси самого глазного яблока. При этом миопия может достигать довольно высоких степеней.
У больных детей также нередко может развиваться синдром «ленивого глаза» — амблиопия. Родителям маленьких пациентов с синдромом Марфана следует быть очень внимательными к состоянию зрения ребенка. Амблиопия зачастую переходит в косоглазие или наоборот.